Quantum computing researchers have completed the largest quantum-assisted protein simulation ever reported, modeling a biologically important system containing more than 12,000 atoms.
The work involved scientists from IBM, RIKEN, and the Cleveland Clinic, who combined quantum computers with advanced supercomputers to study protein-ligand interactions in realistic conditions.
The achievement marks a major step toward practical applications of quantum computing in chemistry and drug research.
The team simulated two protein systems, T4 lysozyme and Trypsin, in aqueous environments. These proteins interact with ligands, which are important in many biological processes and in pharmaceutical research.
READ ALSO: NATO Eyes Cheap Drone Interceptors as Ukraine Challenges US Defense Market Strategy
The largest simulation reached 12,635 atoms and nearly 30,000 molecular orbitals, making it the biggest heterogeneous quantum-classical chemistry calculation to date.
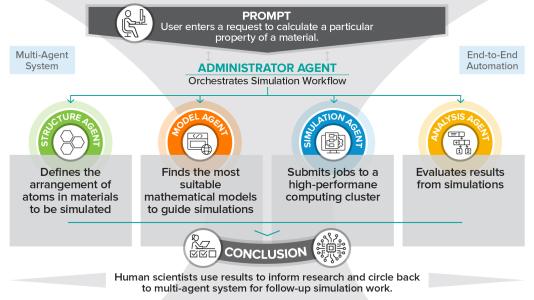
Researchers used a quantum-centric supercomputing workflow that combined quantum processing units, CPUs, and GPUs. The workflow connected two IBM quantum computers with Japan’s Fugaku and Miyabi-G supercomputers. Scientists said the setup allowed different types of processors to work together on separate parts of the same chemistry problem.
The study used up to 94 qubits on two IBM Quantum Heron r2 processors. Researchers ran more than 9,200 quantum circuits over 100 hours and collected 1.3 billion measurement outcomes. According to the research team, this is the most resource-intensive quantum-centric chemistry calculation reported to date.
The new result follows an earlier quantum chemistry milestone achieved only four months ago. In that earlier work, researchers modeled a much smaller 303-atom protein, Trp-cage, using quantum-computing techniques. The latest project expanded the scale by about 40 times while improving accuracy by more than 200 times at a key stage of the workflow.
WATCH ALSO: From Ice to Innovation: US Coast Guard Deploys Drones in Antarctic Mission
Scientists focused on proteins because chemistry fundamentally operates according to quantum mechanics. Traditional computers can model many chemical interactions, but highly accurate simulations become extremely difficult as molecules grow larger. Quantum computers are designed to handle quantum behavior directly, making them promising tools for studying complex chemistry problems.
Dr. Kenneth Merz, from the Cleveland Clinic and the paper’s lead author, said advances in classical computing have slowed compared with earlier decades. He explained that past gains came from faster chips, parallel computing, and graphics processors. Researchers now see quantum computing as a possible path for achieving another major leap in simulation capability.
To manage the enormous complexity of protein calculations, the researchers used a method called wave-function-based embedding (EWF). This approach breaks a large molecule into smaller pieces called clusters. Classical computers handle the simpler parts, while quantum computers handle the more complex quantum interactions.
After solving the clusters separately, the system combines them into a complete molecular model. This strategy reduces the overall computational burden and enables larger simulations. Researchers said the method creates a practical path toward scaling quantum chemistry to much larger biological systems.
The workflow also relied on a technique called sample-based quantum diagonalization(SQD). In chemistry simulations, electrons can arrange themselves in huge numbers of possible configurations. SQD helps the quantum computer identify the most important configurations so classical computers can focus on the most useful data.
READ ALSO: Archaeologists Uncover 5,000-Year-Old Wooden Secret Beneath Stone Island in Scotland
The team introduced a refined version of the method called TrimSQD. Instead of searching one massive configuration space, TrimSQD divides the problem into smaller sections that can be solved independently. Researchers compared the process to sorting puzzle pieces into smaller groups before assembling a complex image.
This refinement improved the system’s ability to separate meaningful quantum states from irrelevant noise. Scientists referred to the useful configurations as livewood, and the less important ones as deadwood. By narrowing the search space, the researchers increased efficiency and improved the quality of the final calculations.
Another major challenge involved handling interactions between electrons across large proteins. Standard methods become extremely expensive as molecular size increases. Researchers said doubling a molecule’s size can increase computational demands by roughly 25 times using conventional approaches.
To solve that problem, the team adopted a linear-scaling method that focused only on nearby interactions. Scientists found that electrons in these proteins mostly interact strongly with atoms within a short distance. By ignoring weak long-range interactions, they dramatically reduced the required computing resources.
This optimization enabled simulations to scale from hundreds of atoms to more than 12,000. Researchers said the improvement was critical for modeling realistic protein systems in liquid environments. Earlier simulations often excluded surrounding water molecules due to computational constraints.
WATCH ALSO: This Microsoft AI Could Change Cancer Treatment Forever
Including water in the simulations added important biological realism. Proteins naturally operate in liquid environments inside living organisms. Modeling proteins in the presence of ligands and water molecules provides researchers with a more accurate picture of molecular behavior.
The international collaboration also demonstrated how future supercomputers may operate. Quantum processors handled specific sampling tasks while classical supercomputers performed large-scale calculations and data processing. Scientists described the project as a practical example of quantum-centric supercomputing architecture.
The work does not yet outperform the best classical chemistry methods across all categories. However, researchers said the study proves that quantum computers can already contribute to meaningful scientific research today. They also expect future quantum hardware to improve rapidly over the next several years.
The methods used in this project are expected to transfer directly to future fault-tolerant quantum computers. IBM plans to develop larger, more reliable quantum systems later this decade, including the IBM Quantum Starling platform. Researchers believe these systems will allow even larger and more accurate chemistry simulations.
The implications extend beyond academic research. Better molecular simulations could help scientists design drugs faster and reduce the need for costly laboratory experiments. The same technology may also advance materials science, energy research, and industrial chemistry.
Researchers are already exploring related applications in biology and materials engineering. Scientists involved in the project said collaboration between quantum researchers and high-performance computing experts will remain essential for future progress.
READ ALSO: B-21 Raider Reveals Hidden Design In First Overhead Image During Secret Trials
The study also highlighted the growing role of hybrid computing systems that combine multiple processor types.
The latest achievement shows how quickly the field is advancing. Only a few years ago, simulations of this scale were considered far beyond the reach of quantum computing systems.
Researchers now expect quantum-assisted chemistry tools to become increasingly important in scientific discovery and pharmaceutical development over the coming decade.